

1
社内資料:サル反復投与毒性試験(2017年3月30日承認、CTD2.6.6.3)
2
社内資料:カニクイザル受胎能及び着床までの初期胚発生に関する試験(2017年3月30日承認、CTD2.6.6.6)
3
社内資料:ウサギ胚・胎児発生に関する試験(2017年3月30日承認、CTD2.6.6.6)
4
Yoshino, T., et al.:Invest New Drugs. 2013;31(4):910-917[ZAL0001]
5
社内資料:母集団薬物動態解析(外国人データ)(2017年3月30日承認、CTD2.7.2.3)
6
社内資料:ラットに静脈内投与後のアフリベルセプト ベータの分布(2017年3月30日承認、CTD2.6.4.4)
7
社内資料:ラットに静脈内投与後のアフリベルセプト ベータの腎排泄(2017年3月30日承認、CTD2.6.4.6)
8
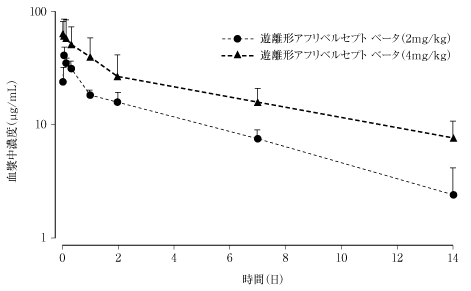
社内資料:国内第2相臨床試験(2017年3月30日承認、CTD2.7.6.2)
9
Van, Cutsem, E., et al.:J. Clin. Oncol. 2012;30(28):3499-3506[ZAL0002]
10
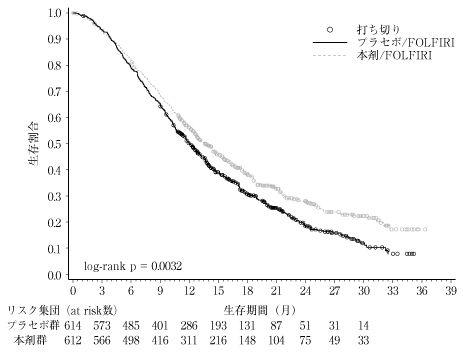
社内資料:海外第3相試験(2017年3月30日承認、CTD2.7.6.2)
11
社内資料:非臨床薬効薬理試験(作用機序)(2017年3月30日承認、CTD2.6.2.1)
12
社内資料:非臨床薬効薬理試験(薬理作用)(2017年3月30日承認、CTD2.6.2.2)

