


1
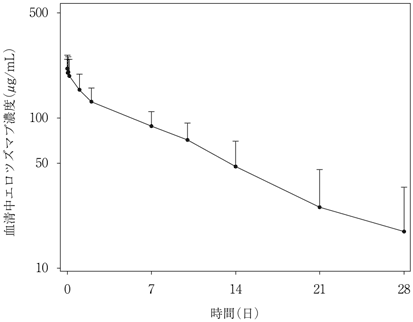
社内資料:海外第1b相臨床試験(C204-007)(平成28年9月28日承認,CTD2.7.2)
2
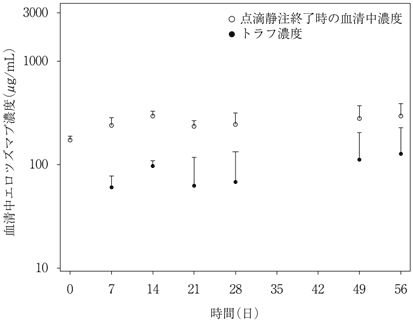
社内資料:国内第1相臨床試験(C204-005)(平成28年9月28日承認,CTD2.7.2)
3
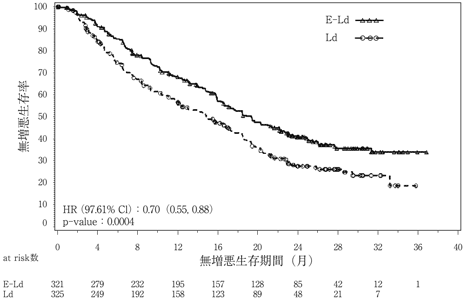
Lonial S, et al.:N Engl J Med. 2015;373(7):621-631.
4
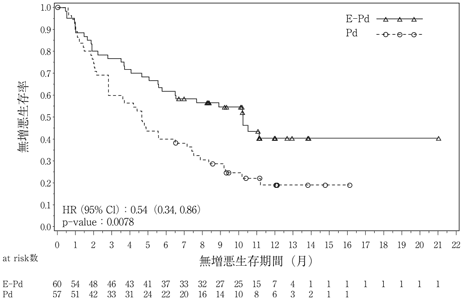
Dimopoulos MA, et al.:N Engl J Med. 2018;379(19):1811-1822.
5
Hsi ED, et al.:Clin Cancer Res. 2008;14(9):2775-2784.
6
Tai YT, et al.:Blood. 2008;112(4):1329-1337.
7
Collins SM, et al.:Cancer Immunol Immunother. 2013;62:1841-1849.

