

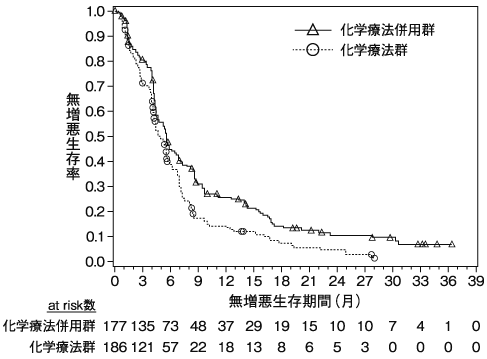
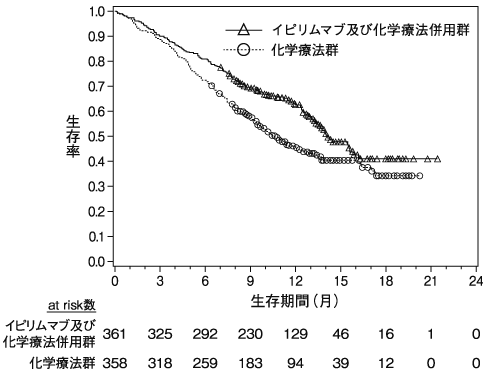
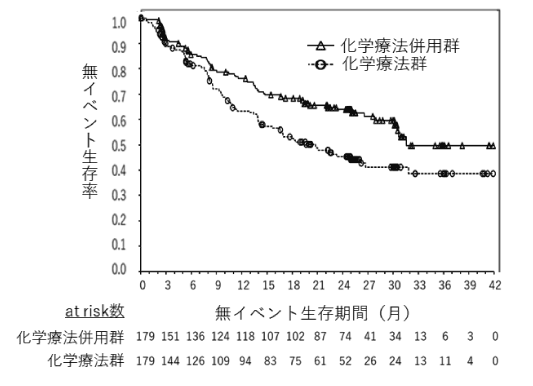
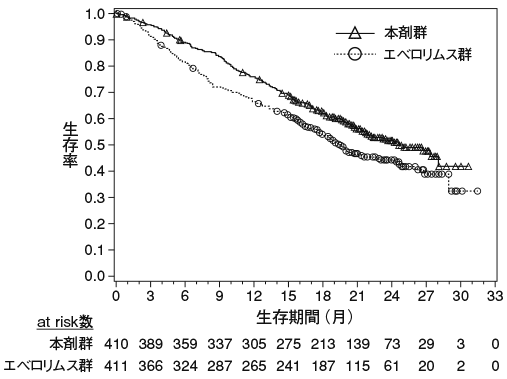
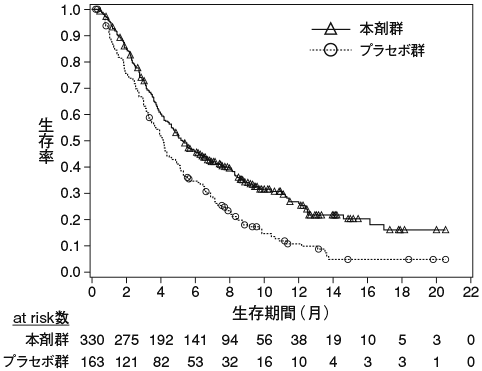
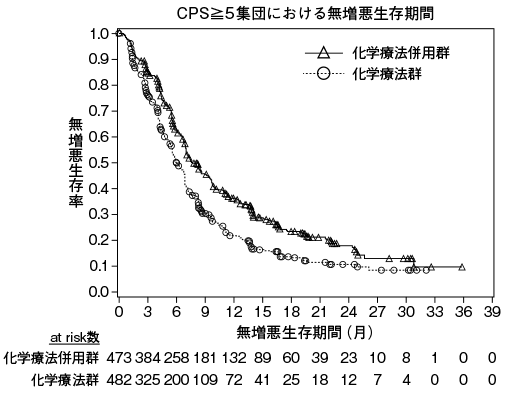
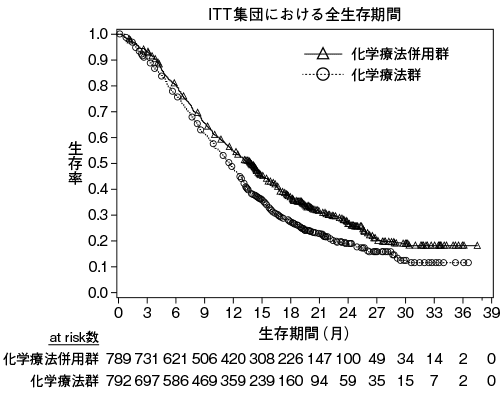
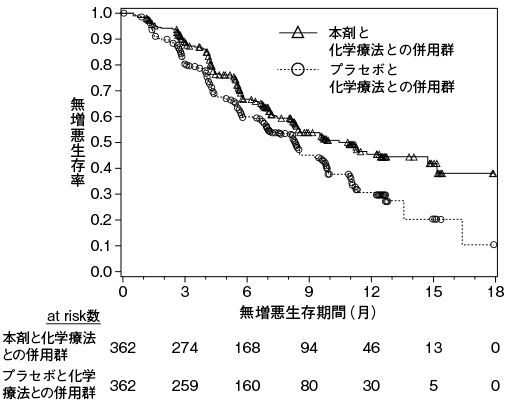
1
オプジーボ点滴静注 適正使用ガイド(単剤療法版・併用療法版)
2
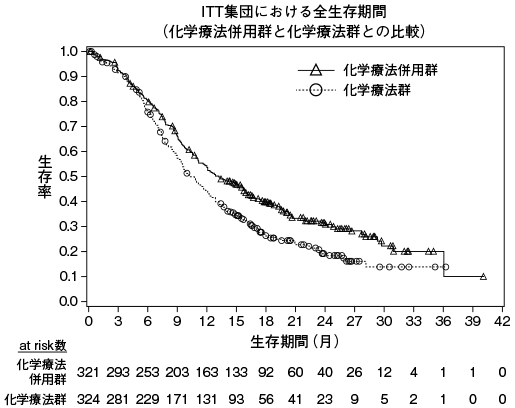
小野薬品工業:海外第Ⅰ相(CA209039)試験成績(社内資料)
3
小野薬品工業:海外第Ⅱ相(CA209205)試験成績(社内資料)
4
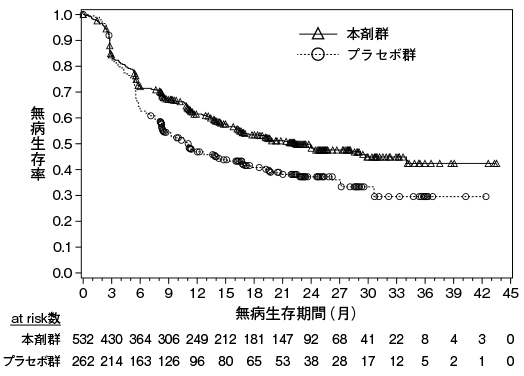
Yamamoto N. et al.:Invest.New Drugs,2017;35:207-216(ONO-4538-01試験)
5
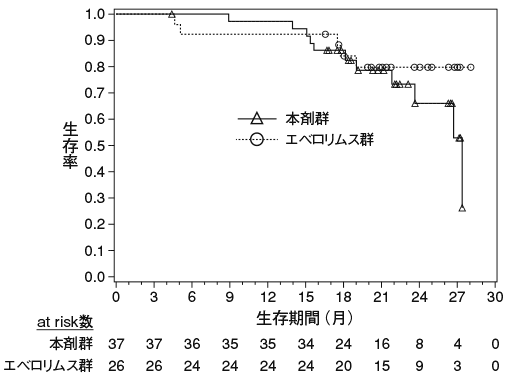
小野薬品工業:国内第Ⅱ相(ONO-4538-02)試験成績(社内資料;2014年7月4日承認、CTD 2.7.6.4)
6
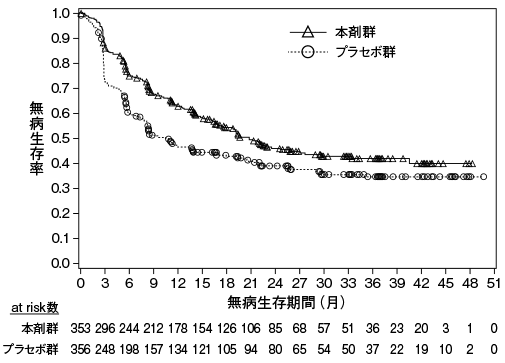
小野薬品工業:国内第Ⅱ相(ONO-4538-08)試験成績(社内資料)
7
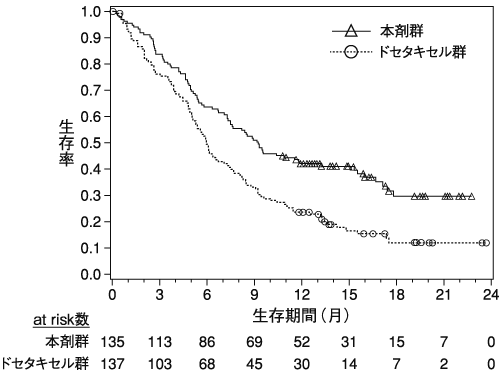
Robert C. et al.:N.Engl.J.Med.,2015;372:320-330(CA209066試験)
8
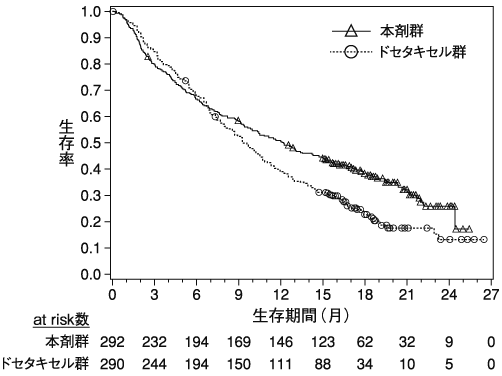
Weber J.S. et al.:Lancet Oncol.,2015;16:375-384(CA209037試験)
9
小野薬品工業:海外第Ⅲ相(CA209037)試験成績(社内資料)
10
小野薬品工業:国内第Ⅱ相(ONO-4538-17)試験成績(社内資料)
11
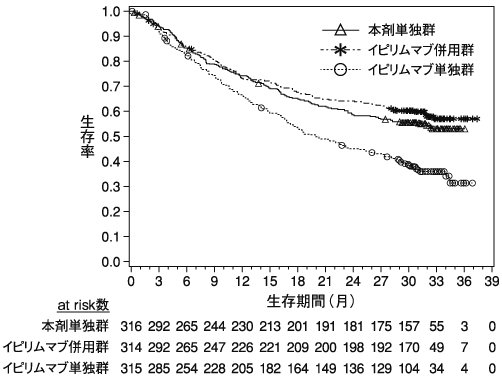
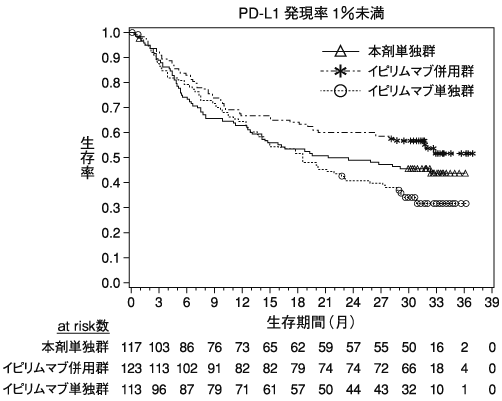
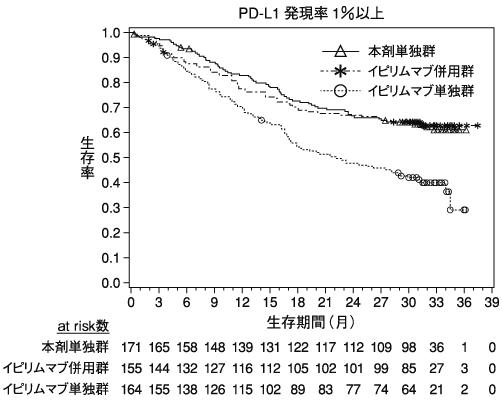
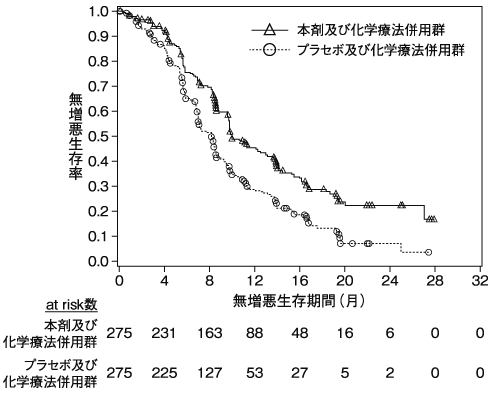
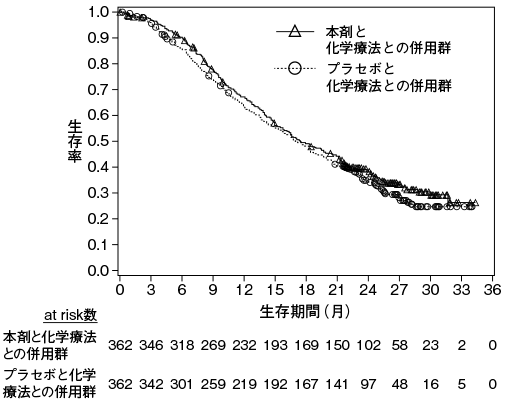
小野薬品工業:海外第Ⅲ相(CA209067)試験成績(社内資料)
12
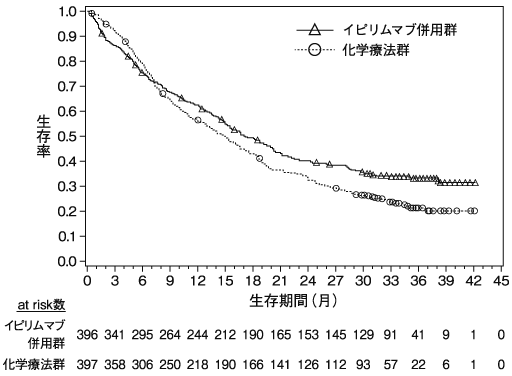
Weber J. et al.:N.Engl.J.Med.,2017;377:1824-1835(ONO-4538-21/CA209238試験)
13
小野薬品工業:国内第Ⅱ相(ONO-4538-05)試験成績(社内資料;2015年12月17日承認、CTD 2.7.6.7)
14
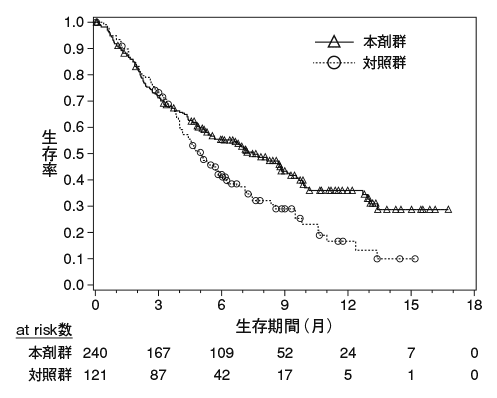
Brahmer J. et al.:N.Engl.J.Med.,2015;373:123-135(CA209017試験)
15
小野薬品工業:国内第Ⅱ相(ONO-4538-06)試験成績(社内資料;2015年12月17日承認、CTD 2.7.6.8)
16
Borghaei H. et al.:N.Engl.J.Med.,2015;373:1627-1639(CA209057試験)
17
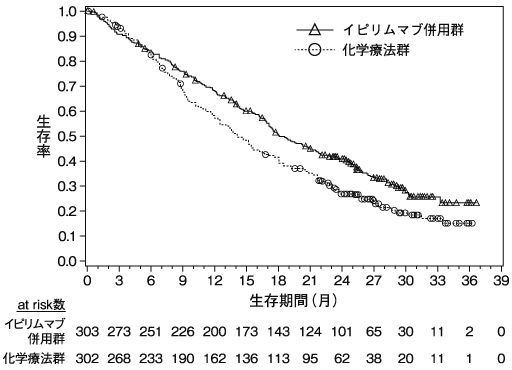
小野薬品工業:国際共同第Ⅲ相(ONO-4538-52)試験成績(社内資料)
18
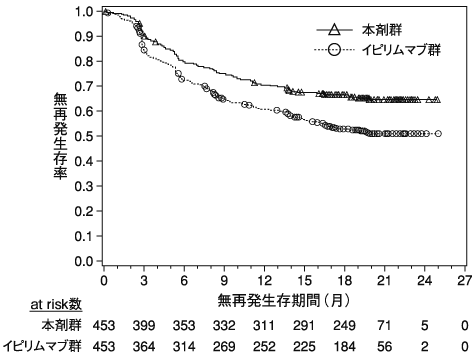
小野薬品工業:国際共同第Ⅲ相(ONO-4538-27/CA209227)試験成績(社内資料)
19
小野薬品工業:国際共同第Ⅲ相(ONO-4538-77/CA2099LA)試験成績(社内資料)
20
小野薬品工業:国際共同第Ⅲ相(ONO-4538-55/CA209816)試験成績(社内資料)
21
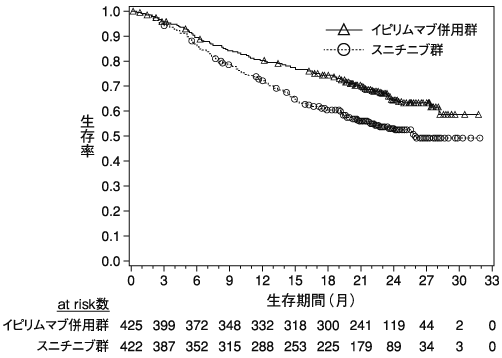
Motzer R.J. et al.:N.Engl.J.Med.,2015;373:1803-1813(ONO-4538-03/CA209025試験)
22
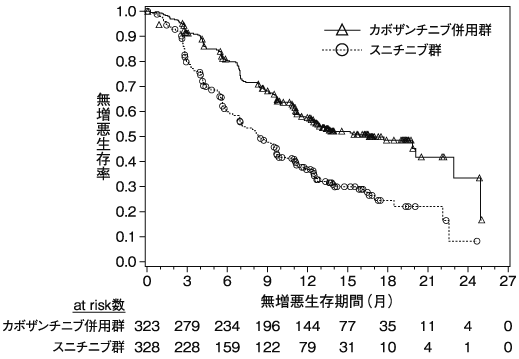
Motzer R.J. et al.:N.Engl.J.Med.,2018;378:1277-1290(ONO-4538-16/CA209214試験)
23
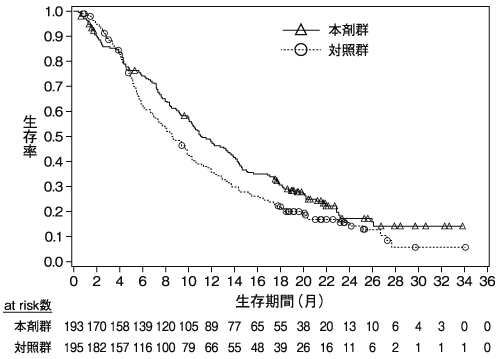
小野薬品工業:国際共同第Ⅲ相(ONO-4538-81/CA2099ER)試験成績(社内資料)
24
小野薬品工業:国内第Ⅱ相(ONO-4538-15)試験成績(社内資料;2016年12月2日承認、CTD 2.7.6.4)
25
Younes A. et al.:Lancet Oncol.,2016;17:1283-1294(CA209205試験)
26
小野薬品工業:国内第Ⅰ相(NCCH1606)試験成績(社内資料;2021年9月27日承認、CTD 2.7.6.1)
27
Ferris R.L. et al.:N.Engl.J.Med.,2016;375:1856-1867(ONO-4538-11/CA209141試験)
28
Kang Y-K. et al.:Lancet,2017;390:2461-2471(ONO-4538-12試験)
29
小野薬品工業:国際共同第Ⅲ相(ONO-4538-44/CA209649)試験成績(社内資料)
30
小野薬品工業:国際共同第Ⅱ/Ⅲ相(ONO-4538-37)試験成績(社内資料)
31
小野薬品工業:国内第Ⅱ相(ONO-4538-41)試験成績(社内資料;2018年8月21日承認、CTD 2.7.6.1)
32
小野薬品工業:国際共同第Ⅲ相(ONO-4538-48/CA209743)試験成績(社内資料)
33
小野薬品工業:国内第Ⅱ相(HCM-002)試験成績(社内資料;2023年11月24日承認、CTD 2.7.6.1)
34
小野薬品工業:海外第Ⅱ相(CA209142)試験成績(社内資料)
35
Overman M.J. et al.:J.Clin.Oncol.,2018;36:773-779(CA209142試験)
36
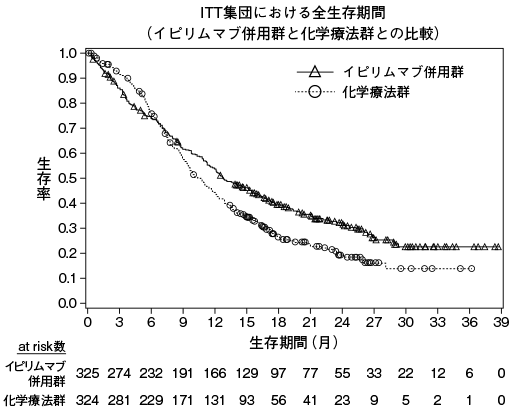
小野薬品工業:国際共同第Ⅲ相(ONO-4538-24/BMS CA209473)試験成績(社内資料)
37
小野薬品工業:国際共同第Ⅲ相(ONO-4538-50/CA209648)試験成績(社内資料)
38
小野薬品工業:国際共同第Ⅲ相(ONO-4538-43/CA209577)試験成績(社内資料)
39
小野薬品工業:国内第Ⅱ相(NM-K2002)試験成績(社内資料;2021年12月24日承認、CTD 2.7.6.1)
40
小野薬品工業:国際共同第Ⅲ相(ONO-4538-33/CA209274)試験成績(社内資料)
41
小野薬品工業:国内第Ⅱ相(KCTR-D014)試験成績(社内資料;2024年2月9日承認、CTD 2.7.6.1)
42
Wang C. et al.:Cancer Immunol.Res.,2014;2:846-856

