

1
Chung CH, et al.:N Engl J Med. 2008;358(11):1109-17.
2
Commins SP, et al.:J Allergy Clin Immunol. 2009;123(2):426-33.
3
Commins SP, et al.:J Allergy Clin Immunol. 2011;127(5):1286-93.
4
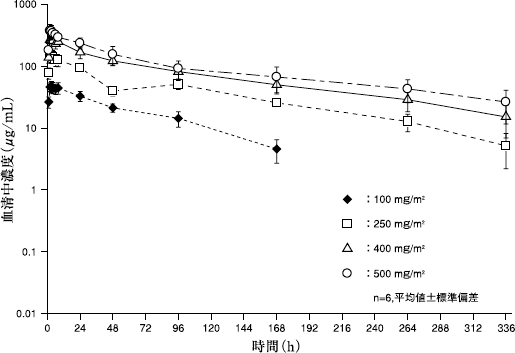
社内資料:国内第I相臨床試験(薬物動態)(2008年7月承認、CTD 2.7.6.2.12)
5
社内資料:母集団薬物動態解析(2008年7月承認、CTD 2.7.2.5.3、CTD 5.3.3.5-1)
6
Delbaldo C, et al.:Eur J Cancer. 2005 Aug;41(12):1739-45.
7
Tahara M, et al.:Jpn J Clin Oncol. 2008 Nov;38(11):762-9
8
社内資料:海外第III相無作為化比較試験(化学療法の前治療歴のない結腸・直腸癌を対象としたフルオロウラシル・ホリナートカルシウム・イリノテカン塩酸塩との併用)EMR62202-013試験
9
Van Cutsem E, et al.:J Clin Oncol. 2011 May 20;29(15):2011-9.
10
Van Cutsem E, et al.:J Clin Oncol. 2015 Mar 1;33(7):692-700.
11
社内資料:海外第III相無作為化比較試験(化学療法の前治療歴のある結腸・直腸癌を対象とした単独投与)(2008年7月承認、CTD 2.7.6-2.36)NCIC CTG CO.17/CA225-025試験
12
Sobrero AF, et al.:J Clin Oncol. 2008 May 10;26(14):2311-9.
13
社内資料:海外第II相無作為化比較試験(化学療法の前治療歴のある結腸・直腸癌を対象としたイリノテカン塩酸塩水和物との併用と単独投与の比較(2008年7月承認、CTD 2.7.6-2.1)EMR62202-007試験
14
Okano S, et al.:Jpn J Clin Oncol. 2013 May;43(5):476-82.
15
Yoshino T, et al.:Jpn J Clin Oncol. 2013 May;43(5):524-31.
16
社内資料:海外第III相無作為化比較試験(局所進行性の頭頸部扁平上皮癌を対象とした放射線療法との併用)(2012年12月承認、CTD 2.7.6.2.2)EMR62202-006/IMCL CP02-9815試験
17
Vermorken JB, et al.:N Engl J Med 2008;359:1116-27
18
Sunada H, et al.:Proc Natl Acad Sci USA. 1986 Jun;83(11):3825-9.
19
Fan Z, et al.:J Biol Chem. 1994 Nov 4;269(44):27595-602.
20
Ciardiello F, et al.:J Natl Cancer Inst. 1998;90(14):1087-94.
21
Morelli MP, et al.:J Cell Physiol. 2006;208(2):344-53.
22
社内資料:抗腫瘍作用(2008年7月承認、CTD 2.6.2.2.2)
23
Luo FR, et al.:Clin Cancer Res. 2005 Aug 1;11(15):5558-65.
24
社内資料:抗腫瘍作用(2008年7月承認、CTD 2.6.2.2.2)

