

1
医療上の必要性の高い未承認薬・適応外薬検討会議 公知申請への該当性に係る報告書:トラスツズマブ(遺伝子組換え)HER2過剰発現が確認された乳癌における術前補助化学療法
2
医療上の必要性の高い未承認薬・適応外薬検討会議 公知申請への該当性に係る報告書:トラスツズマブ(遺伝子組換え)HER2過剰発現が確認された乳癌に対する術後補助化学療法としてのA法(1週間間隔投与)の用法・用量の追加
3
医療上の必要性の高い未承認薬・適応外薬検討会議 公知申請への該当性に係る報告書:トラスツズマブ(遺伝子組換え)HER2過剰発現が確認された転移性乳癌について、3週間1回投与の用法・用量の追加
4
動物実験 胎児移行性(2001年4月4日承認、申請資料概要へ.2-2-3)
5
動物実験 乳汁中移行(2001年4月4日承認、申請資料概要へ.2-4-2)
6
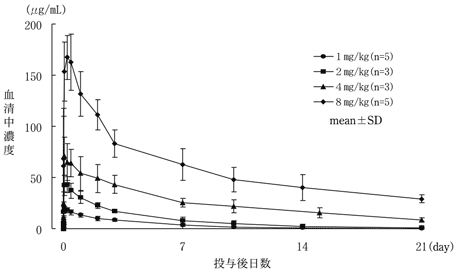
国内第Ⅰ相試験-初回投与時の血中濃度(2001年4月4日承認、申請資料概要へ.3-1-1-2)
7
国内第Ⅰ相試験-反復投与時の血中濃度(2001年4月4日承認、申請資料概要へ.3-1-2-4)
8
社内資料:薬物動態〈HERA試験におけるPKサブスタディ-中間解析結果〉
9
海外第Ⅲ相試験-反復投与(2001年4月4日承認、申請資料概要へ.3-1-2-3)
10
Bruno R, et al. Cancer Chemother Pharmacol. 2005;56:361-9.
11
動物実験 臓器・組織中濃度(2001年4月4日承認、申請資料概要へ.2-2-1)
12
動物実験 代謝物(2001年4月4日承認、申請資料概要へ.2-3-1)
13
国内第Ⅰ相試験-排泄(2001年4月4日承認、申請資料概要へ.3-2-1)
14
動物実験 尿糞中排泄(2001年4月4日承認、申請資料概要へ.2-4-1)
15
国内第Ⅰ相臨床試験(MKC-454-02試験)(2001年4月4日承認、申請資料概要ト.1-1-1-1)
16
海外第Ⅱ相臨床試験(H0551g試験)(2001年4月4日承認、申請資料概要ト.2-3-1)
17
海外第Ⅱ相臨床試験(H0552g試験)(2001年4月4日承認、申請資料概要ト.2-3-2)
18
海外第Ⅲ相試験(H0648g試験)(2001年4月4日承認、申請資料概要ト.2-4-1)
19
海外第Ⅲ相試験(H0649g試験)(2001年4月4日承認、申請資料概要ト.2-4-2)
20
海外臨床試験(H0650g試験)(2001年4月4日承認、申請資料概要ト.2-5-1)
21
海外臨床試験(H0659g試験)(2001年4月4日承認、申請資料概要ト.2-5-2)
22
海外臨床試験(H0693g試験)(2001年4月4日承認、申請資料概要ト.2-5-3)
23
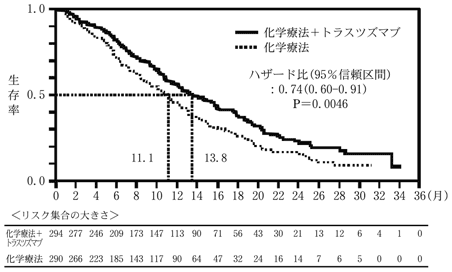
社内資料:臨床成績〈HERA試験-中間解析結果〉
24
Goldhirsch A, et al. Lancet. 2013;382:1021-8.
25
社内資料:臨床成績〈ToGA試験〉
26
社内資料:国内第Ⅱ相試験〈HUON-003-01試験〉
27
社内資料:臨床成績〈TRIUMPH試験〉
28
Lewis GD, et al. Cancer Immunol Immunother. 1993;37:255-63.
29
作用機作〈抗体依存性細胞障害作用(ADCC)〉(2001年4月4日承認、申請資料概要ホ.1-1-1)
30
作用機作〈HER2受容体数抑制作用〉(2001年4月4日承認、申請資料概要ホ.1-1-5)
31
Pietras RJ, et al. Oncogene. 1998;17:2235-49.
32
Baselga J, et al. Cancer Res. 1998;58:2825-31.
33
Fujimoto-Ouchi K, et al. Cancer Chemother Pharmacol. 2007;59:795-805.
34
社内資料:抗腫瘍効果〈ヒト胃癌xenograftモデルにおける抗腫瘍効果の検討〉

