






1
社内資料:ラット胚・胎児発生毒性試験(2019年6月18日承認、CTD2.6.6.6)
2
Nybakken GE, et al.:Leukemia. 2016;30(6):1422-1425
3
社内資料:細菌を用いた復帰突然変異試験(2019年6月18日承認、CTD2.6.6.4)
4
社内資料:トランスジェニックげっ歯類を用いた遺伝子突然変異試験
5
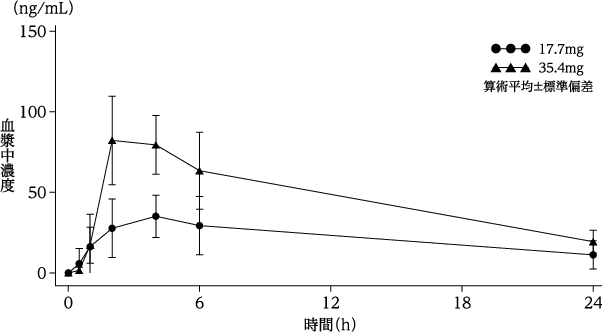
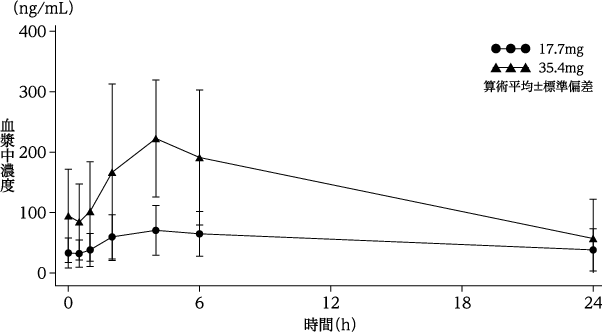
社内資料:初発の急性骨髄性白血病患者を対象とした国内第Ⅰ相試験
6
社内資料:母集団薬物動態解析
7
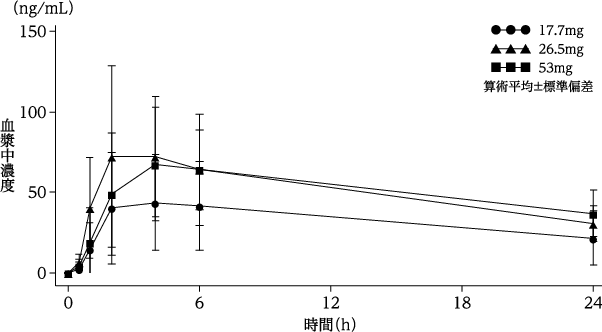
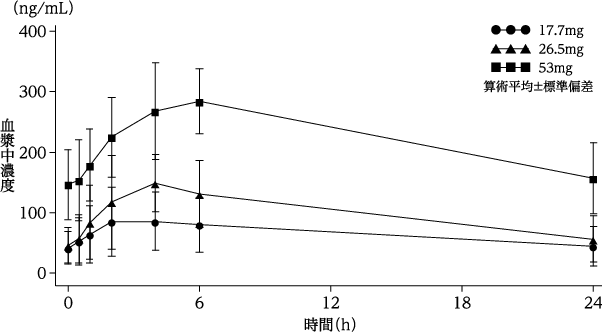
社内資料:再発・難治性急性骨髄性白血病患者を対象とした国内第Ⅰ相試験(2019年6月18日承認、CTD2.7.6.5)
8
社内資料:母集団薬物動態解析(2019年6月18日承認、CTD2.7.2.2)
9
社内資料:外国人における絶対バイオアベイラビリティ試験
10
社内資料:食事の影響試験(2019年6月18日承認、CTD2.7.6.2)
11
社内資料:ヒト血漿蛋白結合率試験(2019年6月18日承認、CTD2.6.4.4)
12
社内資料:ヒト血液/血漿中濃度比試験(2019年6月18日承認、CTD2.6.4.4)
13
社内資料:外国人におけるマスバランス試験(2019年6月18日承認、CTD2.7.6.4)
14
社内資料:CYP分子種同定試験(2019年6月18日承認、CTD2.6.4.5)
15
社内資料:軽度及び中等度肝機能障害者を対象とした薬物動態試験(2019年6月18日承認、CTD2.7.6.7)
16
社内資料:中等度肝機能障害者を対象とした薬物動態試験
17
社内資料:CYP3A阻害剤との薬物相互作用試験(2019年6月18日承認、CTD2.7.6.8)
18
社内資料:エファビレンツとの薬物相互作用試験
19
社内資料:ランソプラゾールとの薬物相互作用試験(2019年6月18日承認、CTD2.7.6.9)
20
社内資料:ダビガトランエテキシラートとの薬物相互作用試験
21
社内資料:トランスポーターを介した輸送評価試験(2019年6月18日承認、CTD2.6.4.3)
22
社内資料:BCRPを介した輸送評価試験
23
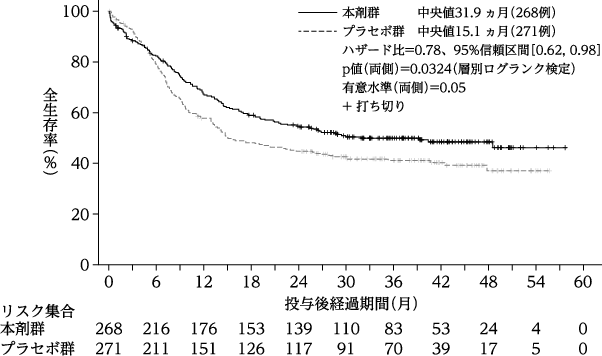
社内資料:FLT3-ITD変異陽性の初発急性骨髄性白血病患者を対象とした国際共同第Ⅲ相試験
24
社内資料:FLT3-ITD変異陽性の再発・難治性急性骨髄性白血病患者を対象とした国内第Ⅱ相試験(2019年6月18日承認、CTD2.7.6.13)
25
Cortes JE, et al.:Lancet Oncol. 2019;20(7):984-997
26
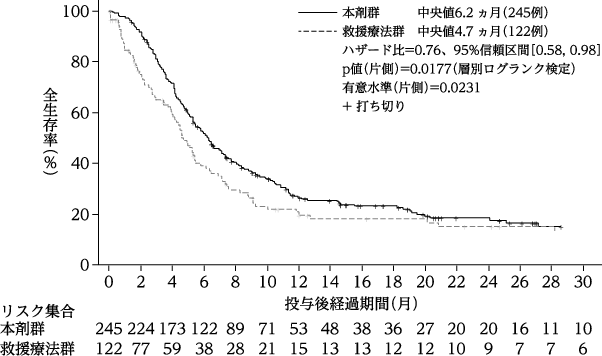
社内資料:FLT3-ITD変異陽性の再発・難治性急性骨髄性白血病患者を対象とした海外第Ⅲ相試験(2019年6月18日承認、CTD2.7.6.12)
27
社内資料:血漿中濃度とQT間隔との関連性(2019年6月18日承認、CTD2.7.2.2)
28
Zarrinkar PP, et al.:Blood. 2009;114(14):2984-2992
29
社内資料:FLT3-ITD変異を有するAML細胞株に対する抗腫瘍作用(2019年6月18日承認、CTD2.6.2.2)
30
社内資料:FLT3-ITD変異を有するAML細胞株に対する化学療法剤との併用による抗腫瘍作用

