


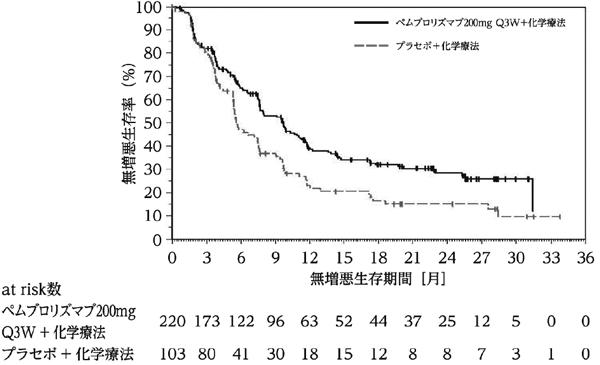
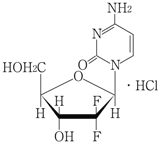
1
膵癌第Ⅰ相試験での薬物動態(ジェムザール注:2001年4月4日承認、申請資料概要 ヘ.1.(1))
2
血漿中濃度(ジェムザール注:2001年4月4日承認、申請資料概要 ヘ.2.(2).1))
3
福岡正博ほか:癌と化学療法 1996;23:1825-1832
4
横山晶ほか:癌と化学療法 1996;23:1681-1688
5
Esumi, Y. et al.:Xenobiotica. 1994;24:957-964
6
尿中・糞中への排泄(ジェムザール注:2001年4月4日承認、申請資料概要 ヘ.2.(2).2))
7
転移性乳癌患者におけるゲムシタビンとパクリタキセル併用投与時の薬物動態(ジェムザール注射用:2010年2月5日承認、審査報告書)
8
福岡正博ほか:癌と化学療法 1996;23:1813-1824
9
Okada, S. et al.:Japanese Journal of Clinical Oncology. 2001;31:7-12
10
第Ⅰ相試験(試験P11D)(ジェムザール注:2001年4月4日承認、申請資料概要ト.1.(1))
11
Rothenberg, M.L. et al.:Annals of Oncology. 1996;7:347-353
12
Burris, H.A. et al.:Journal of Clinical Oncology. 1997;15:2403-2413
13
第Ⅲ相試験(試験JHAY)(ジェムザール注:2001年4月4日承認、申請資料概要ト.1.(3))
14
Okusaka, T. et al.:Cancer Chemotherapy and Pharmacology. 2006;57:647-653
15
Von der Maase, H. et al.:Journal of Clinical Oncology. 2000;18:3068-3077
16
Albain, K.S. et al.:Journal of Clinical Oncology. 2008;26:3950-3957
17
Cortes, J. et al.:Lancet. 2020;396:1817-1828
18
Plunkett, W. et al.:Cancer Research. 1988;48:4024-4031
19
Plunkett, W. et al.:Cancer Research. 1991;51:6110-6117
20
Plunkett, W. et al.:Seminars in Oncology. 1995;22:19-25
21
Plunkett, W. et al.:Purine and Pyrimidine Metabolism in Man VII. 1991;PartA:125-130
22
Plunkett, W. et al.:Molecular Pharmacology. 1990;38:567-572
23
Von Hoff, D.D. et al.:Anti-Cancer Drugs. 1992;3:143-146
24
Peters, G.J. et al.:Purine and Pyrimidine Metabolism in Man VII. 1991;PartA:57-60
25
Bhalla, K. et al.:Gynecologic Oncology. 1992;45:32-39
26
Momparler, R.L. et al.:Anti-Cancer Drugs. 1991;2:49-55
27
Weber, G. et al.:Biochemical and Biophysical Research Communications. 1992;184:551-559
28
Rockwell, S. et al.:Oncology Research. 1992;4:151-155
29
Hertel, L.W. et al.:Cancer Research. 1990;50:4417-4422
30
Plunkett, W, et al.:Cancer Research. 1990;50:3675-3680
31
Braakhuis, B.J.M. et al.:Cancer Research. 1991;51:211-214
32
Kristjansen, P.E.G. et al.:Annals Oncology. 1993;4:157-160
33
藤田昌英ほか:癌と化学療法 1994;21:517-523
34
Peters, G.J. et al.:Seminars in Oncology. 1995;22:72-79
35
Schultz, R.M. et al.:Oncology Research. 1993;5:223-228
36
薬理作用①(ジェムザール注射用:2006年6月15日承認、審査報告書)
37
薬理作用②(ジェムザール注射用:2008年11月25日承認、審査報告書)

