









1
社内資料:ラットにおける胎盤通過性の検討(承認年月日:2013年6月28日、CTD 2.6.4.4.5)
2
社内資料:ラットにおける乳汁移行性の検討(承認年月日:2013年6月28日、CTD 2.6.4.6.6)
3
社内資料:ラットにおけるがん原性試験(承認年月日:2013年6月28日、CTD 2.6.6.5.4)
4
社内資料:マウスにおけるがん原性試験(承認年月日:2013年6月28日、CTD 2.6.6.5.2)
5
社内資料:ラットにおけるクエン酸塩の影響の検討(承認年月日:2013年6月28日、CTD 2.6.6.8.7)
6
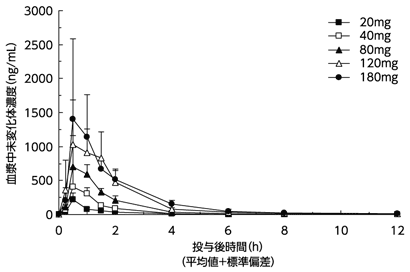
社内資料:健康成人における単回投与試験(承認年月日:2013年6月28日、CTD 2.7.6.3.1)
7
社内資料:健康成人における反復投与試験(承認年月日:2013年6月28日、CTD 2.7.6.3.2)
8
社内資料:健康成人における食事の影響試験(承認年月日:2013年6月28日、CTD 2.7.6.1.1)
9
社内資料:ヒト血漿を用いた蛋白結合に関する検討(承認年月日:2013年6月28日、CTD 2.6.4.4.3)
10
Omura K, et al.:Drug Metab Dispos. 2007;35(12):2143-2148
11
社内資料:代謝酵素活性に対する阻害作用の検討(承認年月日:2013年6月28日、CTD 2.6.4.7.2)
12
社内資料:薬物トランスポーターに対する阻害作用の検討(承認年月日:2013年6月28日、CTD 2.6.4.7.4)
13
社内資料:マスバランス試験(承認年月日:2013年6月28日、CTD 2.7.6.3.5)
14
社内資料:腎機能低下被験者における薬物動態試験(承認年月日:2013年6月28日、CTD 2.7.6.3.6)
15
社内資料:高齢者における薬物動態試験(承認年月日:2013年6月28日、CTD 2.7.6.3.7)
16
社内資料:女性高齢者における薬物動態試験(承認年月日:2013年6月28日、CTD 2.7.6.3.8)
17
社内資料:ワルファリンとの相互作用試験(承認年月日:2013年6月28日、CTD 2.7.6.3.10)
18
Hosoya T, et al.:Clin Rheumatol. 2017;36(3):649-656
19
社内資料:第Ⅱ相(Ⅱb)試験(承認年月日:2013年6月28日、CTD 2.7.6.5.3)
20
Hosoya T, et al.:J Clin Pharm Ther. 2016;41(3):290-297
21
社内資料:第Ⅲ相アロプリノール比較試験(承認年月日:2013年6月28日、CTD 2.7.6.5.4)
22
Hosoya T, et al.:Clin Drug Investig. 2018;38(12):1135-1143
23
Hosoya T, et al.:Clin Exp Nephrol. 2014;18(6):876-884
24
社内資料:第Ⅲ相CKD試験(承認年月日:2013年6月28日、CTD 2.7.6.5.7)
25
社内資料:キサンチン酸化還元酵素阻害試験(承認年月日:2013年6月28日、CTD 2.6.2.2.1)
26
社内資料:アルデヒドオキシダーゼ及びプリン・ピリミジン代謝酵素阻害試験(承認年月日:2013年6月28日、CTD 2.6.2.2.3)
27
社内資料:マウスにおける尿酸低下試験(承認年月日:2013年6月28日、CTD 2.6.2.2.6)
28
社内資料:ラットにおける尿酸低下試験(承認年月日:2013年6月28日、CTD 2.6.2.2.5)
29
社内資料:チンパンジーにおける作用検討試験(承認年月日:2013年6月28日、CTD 2.6.2.2.7)

