










1
社内報告: 生殖発生毒性(ラット). 2010.(2011年1月21日承認、CTD2.6.6.6)
2
Lehtisalo M, et al.: Clin Transl Sci. 2020; 13(6): 1236-43.
3
社内報告: がん原性(マウス、ラット). 2010.(2011年1月21日承認、CTD2.6.6.5)
4
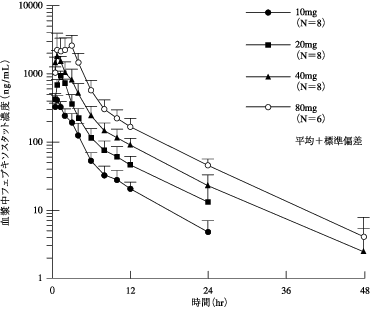
社内報告: 血漿中濃度及び排泄(健康成人、単回). 2010.(2011年1月21日承認、CTD2.7.6.1)
5
社内報告: 血漿中濃度(健康成人、単回). 2010.(2011年1月21日承認、CTD2.7.6.5)
6
社内報告: 血漿中濃度(健康成人、反復). 2010.(2011年1月21日承認、CTD2.7.6.8)
7
社内報告: 血漿中濃度(高尿酸血症患者、反復). 2010.(2011年1月21日承認、CTD2.7.6.31)
8
社内報告: 母集団薬物動態解析(健康成人、腎機能低下患者及び小児高尿酸血症患者). 2023.(2023年6月26日承認、審査報告書)
9
Mukoyoshi M, et al.: Xenobiotica. 2008; 38(5): 496-510.
10
Grabowski BA, et al.: J Clin Pharmacol. 2011; 51(2): 189-201.
11
社内報告: 代謝(ヒト). 2010.(2011年1月21日承認、CTD2.7.2.2.1)
12
社内報告: 血漿中濃度(腎機能低下患者、反復). 2010.(2011年1月21日承認、CTD2.7.6.13)
13
Mayer MD, et al.: Am J Ther. 2005; 12(1): 22-34.
14
Khosravan R, et al.: J Clin Pharmacol. 2006; 46(1): 88-102.
15
Khosravan R, et al.: J Clin Pharmacol. 2008; 48(9): 1014-24.
16
Khosravan R, et al.: Br J Clin Pharmacol. 2008; 65(3): 355-63.
17
社内報告: 薬物相互作用(コルヒチン). 2010.(2011年1月21日承認、CTD2.7.6.18、2.7.6.19)
18
Khosravan R, et al.: J Clin Pharmacol. 2006; 46(8): 855-66.
19
社内報告: 薬物相互作用(デシプラミン). 2010.(2011年1月21日承認、CTD2.7.6.16)
20
社内報告: 薬物相互作用(ワルファリン). 2010.(2011年1月21日承認、CTD2.7.6.23、2.7.6.24)
21
Grabowski BA, et al.: Br J Clin Pharmacol. 2010; 70(1): 57-64.
22
社内報告: 薬物相互作用(テオフィリン). 2010.(2011年1月21日承認、CTD2.7.6.25)
23
社内報告: 薬物相互作用(ロシグリタゾン). 2010.(2011年1月21日承認、CTD2.7.6.26)
24
社内報告: プラセボ対照無作為化二重盲検用量反応比較試験(痛風を含む高尿酸血症患者). 2010.(2011年1月21日承認、CTD2.7.6.33)
25
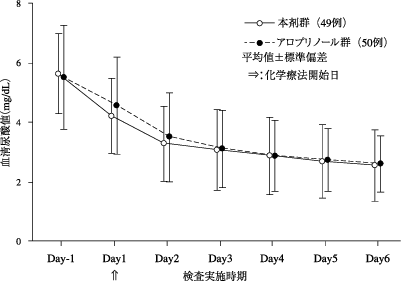
社内報告: アロプリノール対照無作為化二重盲検比較試験(痛風を含む高尿酸血症患者). 2010.(2011年1月21日承認、CTD2.7.6.35a)
26
社内報告: 長期投与試験(痛風を含む高尿酸血症患者). 2010.(2011年1月21日承認、CTD2.7.6.40)
27
社内報告: 国内第Ⅱ相試験(痛風を含む高尿酸血症の小児患者). 2023.(2023年6月26日承認、CTD2.7.6.8)
28
社内報告: 国内第Ⅱ相継続投与試験(痛風を含む高尿酸血症の小児患者). 2023.(2023年6月26日承認、CTD2.7.6.9)
29
Tamura K, et al.: Int J Clin Oncol. 2016; 21(5): 996-1003.
30
Takano Y, et al.: Life Sci. 2005; 76(16):1835-47.
31
社内報告: 血中及び尿中尿酸低下作用(ラット). 2010.(2011年1月21日承認、CTD2.6.2.2.2)

