




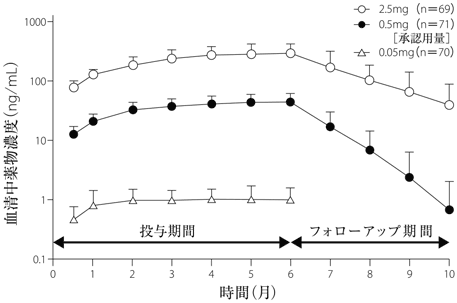
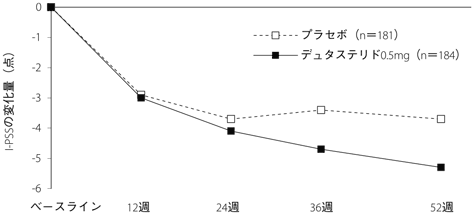
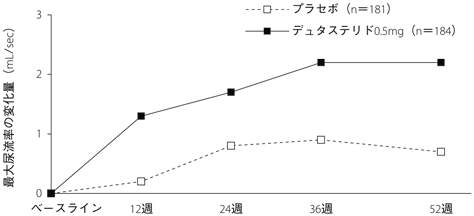
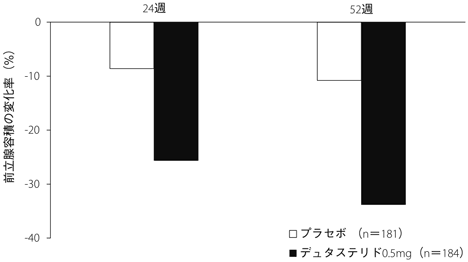
1
Andriole GL,et al.:N Engl J Med.2010;362:1192-1202
2
Theoret MR,et al.:N Engl J Med.2011;365:97-99
3
Akaza H,et al.:Jpn J Clin Oncol.2011;41:417-423
4
塚本泰司ほか:泌尿紀要.2009;55:209-214
5
Tsukamoto T,et al.:Int J Urol.2009;16:745-750
6
Tian G,et al.:Biochemistry.1995;34:13453-13459
7
Bramson HN,et al.:J Pharmacol Exp Ther.1997;282:1496-1502

