


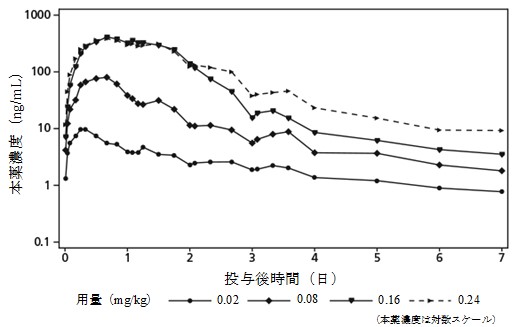

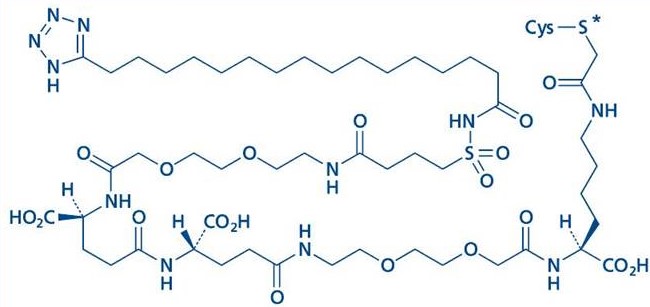
1
社内資料:第I相臨床試験(NN8640-3915)(2021年1月22日承認 CTD2.7.2.3)
2
社内資料:第I相臨床試験(NN8640-3947)(2021年1月22日承認 CTD2.7.2.3)
3
社内資料:第I相臨床試験(NN8640-4042)(202x年xx月xx日承認 CTD2.7.2.3)
4
社内資料:2021年1月22日承認 CTD2.7.2.3.1.9
5
社内資料:202x年xx月xx日承認 CTD2.7.2.3.1.6
6
社内資料:第I相臨床試験(NN8640-4237)(2021年1月22日承認 CTD2.7.2.3)
7
社内資料:第I相臨床試験(NN8640-4297)(2021年1月22日承認 CTD2.7.2.3)
8
社内資料:第I相臨床試験(NN8640-4298)(2021年1月22日承認 CTD2.7.2.3)
9
社内資料:第III相臨床試験(NN8640-4054)(2021年1月22日承認CTD2.7.3.2及び2.7.3.3)
10
社内資料:第III相臨床試験(NN8640-4054)(2021年1月22日承認 CTD2.7.6.6)
11
社内資料:第III相臨床試験(NN8640-4054)(2021年1月22日承認 CTD2.7.3.5)
12
社内資料:第III相臨床試験(NN8640-4054)(2021年1月22日承認 CTD2.7.6.7)
13
社内資料:第III相臨床試験(NN8640-4244)(2021年1月22日承認CTD2.7.3.2及び2.7.3.3)
14
社内資料:第III相臨床試験(NN8640-4244)(2021年1月22日承認 CTD2.7.6.9)
15
社内資料:第III相臨床試験(NN8640-4263)(202x年xx月xx日承認 CTD2.7.3.2、2.7.3.3、2.7.3.5、2.7.6.6及び2.7.6.7)
16
社内資料:2021年1月22日承認 CTD2.4.1.1
17
社内資料:2021年1月22日承認 CTD2.6.2.1

