

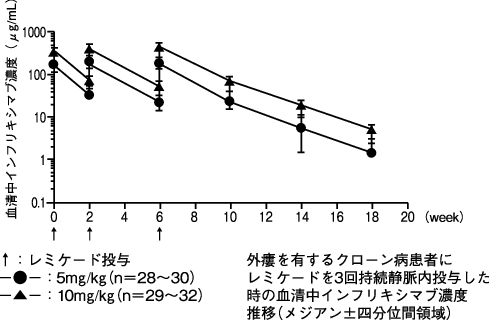
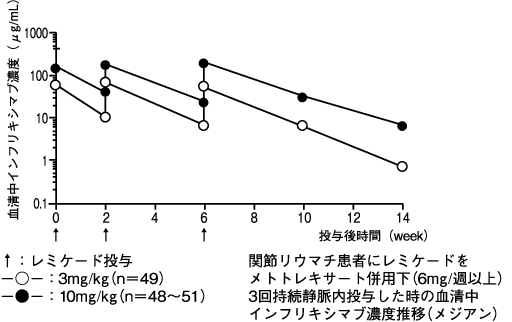
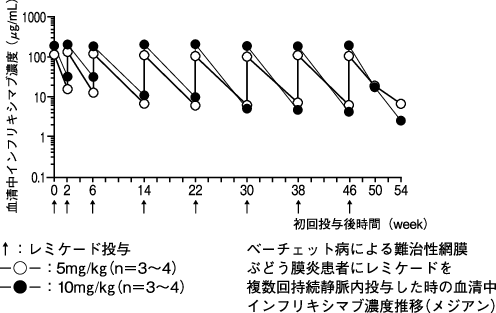
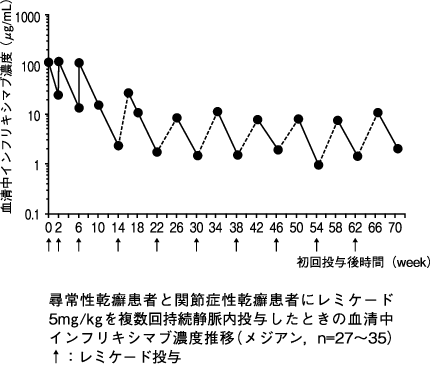
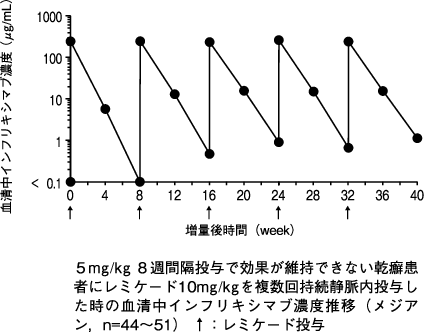
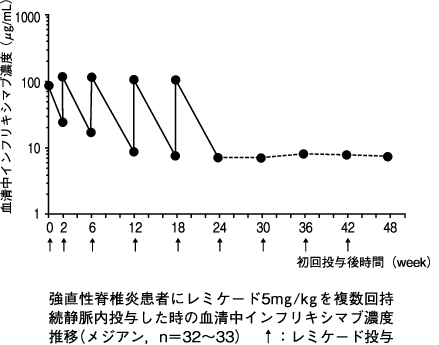
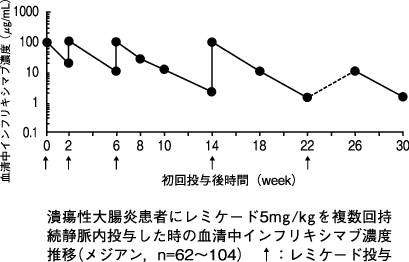
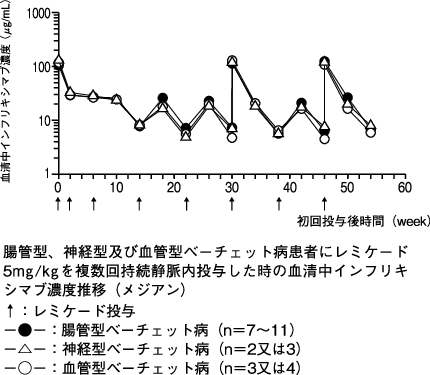
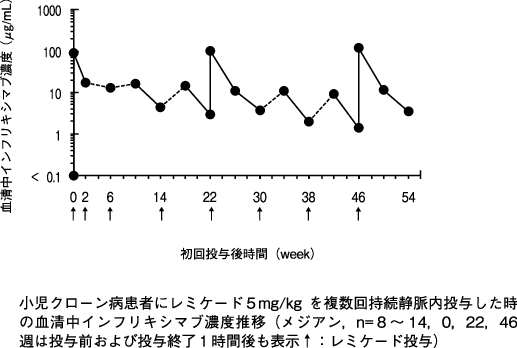
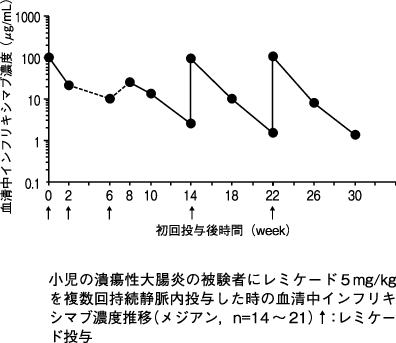
1
Westhovens R, et al.:Arthritis Rheum. 2006;54(4):1075-1086
2
Asakura H, et al.:J Gastroenterol Hepatol. 2001;16(7):763-769
3
Targan SR, et al.:N Engl J Med. 1997;337(15):1029-1035
4
Hanauer SB, et al.:Lancet. 2002;359:1541-1549
5
Present DH, et al.:N Engl J Med. 1999;340(18):1398-1405
6
Sands BE, et al.:N Engl J Med. 2004;350(9):876-885
7
Lipsky PE, et al.:N Engl J Med. 2000;343(22):1594-1602
8
Antoni C, et al.:Ann Rheum Dis. 2005;64(8):1150-1157
9
van der Heijde D, et al.:Arthritis Rheum. 2007;56(8):2698-2707
10
van der Heijde D, et al.:Arthritis Rheum. 2005;52(2):582-591
11
Rutgeerts P, et al.:N Engl J Med. 2005;353(23):2462-2476
12
Scallon BJ, et al.:Cytokine. 1995;7(3):251-259
13
Siegel SA, et al.:Cytokine. 1995;7(1):15-25

