




1
社内資料:ラットにおける乳汁排泄(承認年月日:2012.6.29、CTD 2.6.4.6)
2
社内資料:健康成人における血中濃度(第Ⅰ相臨床試験 単回投与)(承認年月日:2012.6.29、CTD 2.7.6.3)
3
社内資料:健康成人における血中濃度(食事の影響)(承認年月日:2012.6.29、CTD 2.7.6.2)
4
社内資料:ラットにおける組織分布(承認年月日:2012.6.29、CTD 2.6.4.4)
5
社内資料:代謝(承認年月日:2012.6.29、CTD 2.6.4.5)
6
社内資料:マスバランス試験(承認年月日:2012.6.29、CTD 2.7.6.1)
7
社内資料:マスバランス試験(承認年月日:2012.6.29、CTD 2.7.2.2)
8
社内資料:薬物動態試験(腎機能障害患者)(承認年月日:2012.6.29、CTD 2.7.6.5)
9
社内資料:薬物動態試験(肝機能障害患者)(承認年月日:2012.6.29、CTD 2.7.6.6)
10
社内資料:第Ⅲ相比較試験(承認年月日:2012.6.29、CTD 2.7.6.8)
11
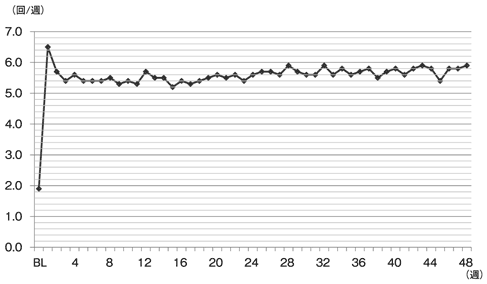
社内資料:第Ⅲ相長期投与試験(承認年月日:2012.6.29、CTD 2.7.6.14)
12
社内資料:オピオイド誘発性腸管機能不全患者を対象とした海外第Ⅲ相比較試験(結果概要)
13
社内資料:アミティーザカプセル12μgに関する資料(治験総括報告書)(承認年月日:2018.9.21、CTD 5.3.1.2)
14
Cuppoletti J,et al.:Am J Physiol Cell Physiol. 2004;287:C1173
15
社内資料:腸管内輸送への影響(承認年月日:2012.6.29、CTD 2.6.2.2)
16
社内資料:ラットでの腸液分泌に対する影響(承認年月日:2012.6.29、CTD 2.6.2.2)
17
社内資料:ラットの腸管内への水分泌に対する影響(承認年月日:2012.6.29、CTD 2.6.2.2)
18
社内資料:腸液中及び血清中の電解質への影響(承認年月日:2012.6.29、CTD 2.6.2.2)

