



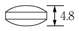
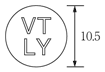
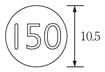
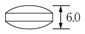
1
社内資料:生殖発生毒性試験(2010年4月16日承認、CTD2.6.6.6)
2
社内資料:健康授乳婦における薬物動態
3
社内資料:幼若動物を用いた毒性試験(2010年4月16日承認、CTD2.6.6.6)
4
社内資料:がん原性試験(2010年4月16日承認、CTD2.6.6.5)]
5
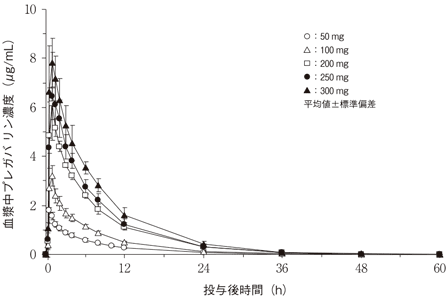
社内資料:健康成人における薬物動態(単回投与)(2010年4月16日承認、CTD2.7.2.2.2.1.1)
6
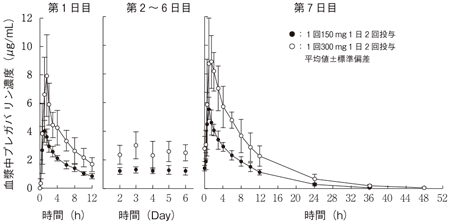
社内資料:健康成人における薬物動態(反復投与)(2010年4月16日承認、CTD2.7.2.2.2.1.1)
7
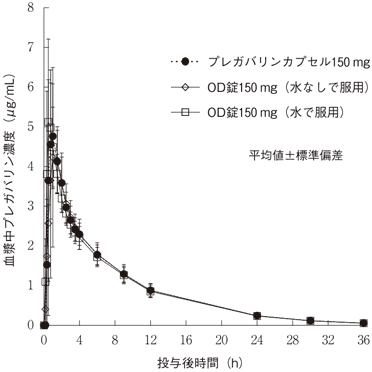
社内資料:生物学的同等性試験
8
社内資料:食事の影響(2010年4月16日承認、CTD2.7.1.2.2)
9
社内資料:放射性標識体投与時の薬物動態及び代謝(2010年4月16日承認、CTD2.7.2.2.1.6)
10
社内資料:血漿蛋白結合(2010年4月16日承認、CTD2.7.2.2.1.5)
11
社内資料:ヒトcytochrome P450に対する阻害作用(2010年4月16日承認、CTD2.7.2.2.1.3)
12
社内資料:高齢者における薬物動態(2010年4月16日承認、CTD2.7.2.2.2.2.1)
13
社内資料:腎機能障害患者及び血液透析患者における薬物動態(2010年4月16日承認、CTD2.7.2.2.2.2.2)
14
社内資料:健康被験者、帯状疱疹後神経痛患者、糖尿病性末梢神経障害に伴う疼痛を有する患者及び線維筋痛症患者における母集団薬物動態(2010年10月27日承認CTD2.7.2.3.5.1、2012年6月22日承認CTD2.7.2.3.1、2.7.2.3.4)
15
社内資料:ガバペンチンとの薬物相互作用(2010年4月16日承認、CTD2.7.2.2.2.3.5)
16
社内資料:経口避妊薬との薬物相互作用(2010年4月16日承認、CTD2.7.2.2.2.3.6)
17
社内資料:ロラゼパムとの薬物相互作用(2010年4月16日承認、CTD2.7.2.2.2.4.1)
18
社内資料:オキシコドンとの薬物相互作用(2010年4月16日承認、CTD2.7.2.2.2.4.2)
19
社内資料:エタノールとの薬物相互作用(2010年4月16日承認、CTD2.7.2.2.2.4.3)
20
Brodie MJ,et al.:Epilepsia. 2005;46(9):1407-1413
21
小川節郎ほか:日本ペインクリニック学会誌. 2010;17(2):141-152
22
社内資料:国内第Ⅲ相検証試験(糖尿病性末梢神経障害に伴う疼痛)(2010年10月27日承認、CTD2.7.3.3)
23
社内資料:国内第Ⅲ相検証試験(線維筋痛症)(2012年6月22日承認、CTD2.7.3.3)
24
社内資料:国内長期投与試験(帯状疱疹後神経痛)(2010年4月16日承認、CTD2.7.3.5.1)
25
社内資料:国内長期投与試験(糖尿病性末梢神経障害に伴う疼痛)(2010年10月27日承認、CTD2.7.3.5.2)
26
社内資料:国内長期投与試験(線維筋痛症)(2012年6月22日承認、CTD2.7.3.5.1)
27
社内資料:国内長期投与試験(脊髄損傷後疼痛、脳卒中後疼痛、多発性硬化症に伴う疼痛)(2013年2月28日承認、CTD2.7.3.5.1)
28
社内資料:国際共同第Ⅲ相試験(脊髄損傷後疼痛)(2013年2月28日承認、CTD2.7.3.3)
29
社内資料:外国第Ⅱ相及び第Ⅲ相プラセボ対照試験(帯状疱疹後神経痛)(2010年4月16日承認、CTD2.7.3.3)
30
社内資料:外国長期投与試験(帯状疱疹後神経痛)(2010年4月16日承認、CTD2.7.3.5.2)
31
Bauer CS,et al.:J Neurosci. 2009;29(13):4076-4088
32
Fink K,et al.:Neuropharmacology. 2002;42(2):229-236
33
Maneuf YP,et al.:Pain. 2001;93(2):191-196
34
Tanabe M,et al.:J Neurosci Res. 2008;86(15):3258-3264
35
Bee LA,et al.:Pain. 2008;140(1):209-223
36
Field MJ,et al.:Pain. 1999;83(2):303-311
37
Field MJ,et al.:Pain. 1999;80(1-2):391-398
38
Tanabe M,et al.:Eur J Pharmacol. 2009;609(1-3):65-68
39
社内資料:慢性筋骨格系疼痛モデルにおける薬効薬理試験(2012年6月22日承認、CTD2.6.2.2)
40
Field MJ,et al.:Br J Pharmacol. 1997;121(8):1513-1522
41
Field MJ,et al.:J Pharmacol Exp Ther. 1997;282(3):1242-1246

