



1
社内資料:ラット乳汁移行性試験(2018年1月19日承認、CTD2.6.5.10)
2
Stone, M. et al.:BMJ.2009;339:b2880.
3
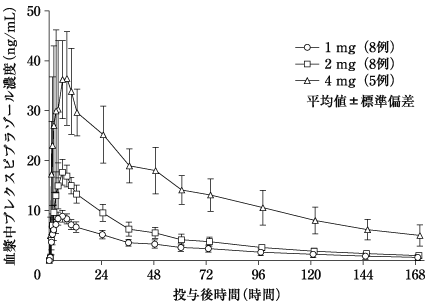
社内資料:国内単回投与試験(2018年1月19日承認、CTD2.7.6.3)
4
社内資料:統合失調症患者を対象とした国内反復投与試験(2018年1月19日承認、CTD2.7.6.3)
5
社内資料:食事の影響試験(2018年1月19日承認、CTD2.7.6.2)
6
社内資料:静注液と錠剤の絶対的バイオアベイラビリティ試験(2018年1月19日承認、CTD2.7.1.2)
7
社内資料:蛋白結合試験(2018年1月19日承認、CTD2.7.2.2)
8
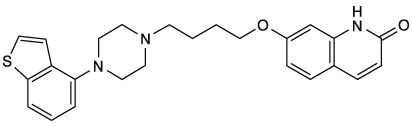
社内資料:推定代謝経路(2018年1月19日承認、CTD2.7.2.2)
9
社内資料:統合失調症患者又は統合失調感情障害患者を対象とした反復投与試験(2018年1月19日承認、CTD2.7.6.3)
10
社内資料:CYP阻害剤併用投与による薬物相互作用試験(2018年1月19日承認、CTD2.7.6.3)
11
社内資料:単回投与時の薬物動態、マスバランス、食事の影響試験(2018年1月19日承認、CTD2.7.6.3)
12
社内資料:腎機能障害患者での薬物動態試験(2018年1月19日承認、CTD2.7.6.3)
13
社内資料:肝機能障害患者での薬物動態試験(2018年1月19日承認、CTD2.7.6.3)
14
社内資料:年齢及び性別の薬物動態への影響(2018年1月19日承認、CTD2.7.6.3)
15
社内資料:リファンピシン併用投与による薬物相互作用試験(2018年1月19日承認、CTD2.7.6.3)
16
社内資料:活性炭併用投与による薬物相互作用試験(2018年1月19日承認、CTD2.7.6.3)
17
社内資料:生理学的薬物速度論(PBPK)モデル解析(2023年12月22日承認、CTD2.7.2.3)
18
社内資料:統合失調症患者を対象とした国内プラセボ対照二重盲検試験(2018年1月19日承認、CTD2.7.6.5)
19
社内資料:統合失調症患者を対象とした海外プラセボ対照二重盲検試験1(2018年1月19日承認、CTD2.7.6.5)
20
社内資料:統合失調症患者を対象とした海外プラセボ対照二重盲検試験2(2018年1月19日承認、CTD2.7.6.5)
21
社内資料:統合失調症患者を対象とした国内長期投与試験(2018年1月19日承認、CTD2.7.6.5)
22
社内資料:うつ病・うつ状態に対する短期試験(2023年12月22日承認、CTD2.7.6.2)
23
Maeda, K. et al.:J Pharmacol Exp Ther.2014;350(3):589-604.
24
Maeda, K. et al.:J Pharmacol Exp Ther.2014;350(3):605-614.
25
社内資料:うつ症状関連の動物モデルにおける改善作用(2023年12月22日承認、CTD2.6.2.2)

