




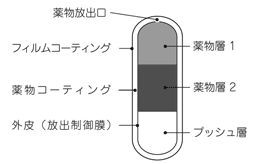

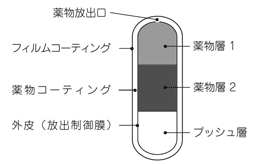

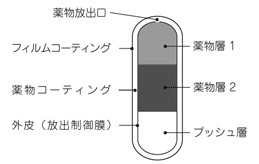
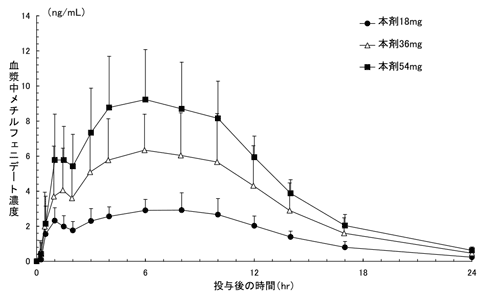
1
Hackett LP, et al.: Ann Pharmacother. 2006 ; 40 : 1890-1891
2
Spigset O, et al.: Am J Psychiatry. 2007; 164 : 348
3
Popper CW.: J Child Adolesc Psychopharmacol. 1995 ; 5 : 157-166
4
National Toxicology Program. 1995 ; TR No.439
5
社内資料:安藤隆康:健康成人におけるコンサータ錠の薬物動態の検討(JNS001-JPN-01)(2013年12月20日承認、CTD 2.7.6.8)
6
社内資料:健康成人におけるコンサータ錠の薬物動態の検討(CONCERTA NAP1003)(2013年12月20日承認、CTD 2.7.6.3)
7
社内資料:安藤隆康:AD/HD患児におけるコンサータ錠の薬物動態の検討(JNS001-JPN-03-1)(2007年10月26日承認、CTD 2.7.6.12)
8
社内資料:コンサータ錠の薬物動態に及ぼす食事の影響(2007年10月26日承認、CTD 2.7.6.13)
9
社内資料:コンサータ錠の薬物動態に及ぼす食事の影響(C-A-002)(2013年12月20日承認、CTD 2.7.6.1)
10
Hungund BL, et al.: Br J Clin Pharmacol. 1979 ; 8 : 571-576
11
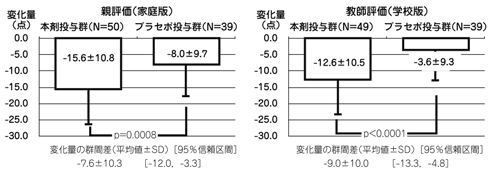
社内資料:安藤隆康:コンサータ錠の第Ⅲ相試験成績(JNS001-JPN-03-2)(2007年10月26日承認、CTD 2.7.6.12)
12
社内資料:安藤隆康:コンサータ錠の長期投与試験成績(JNS001-JPN-04)(2007年10月26日承認、CTD 2.7.6.17)
13
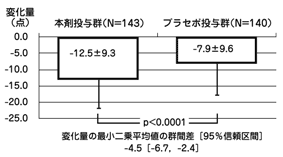
社内資料:高橋長秀:コンサータ錠の第Ⅲ相試験成績(JNS001-JPN-A01)(2013年12月20日承認、CTD 2.7.6.8)
14
社内資料:高橋長秀:コンサータ錠の長期投与試験成績(JNS001-JPN-A02)(2013年12月20日承認、CTD 2.7.6.12)
15
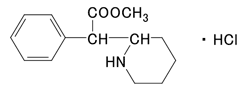
社内資料:塩酸メチルフェニデートの作用機序
16
Ueno KI, et al.: Behav Pharmacol. 2002 ; 13 : 1-13
17
Kula NS, et al.: Eur J Pharmacol. 1999 ; 385 : 291-294

