


 P1
P1
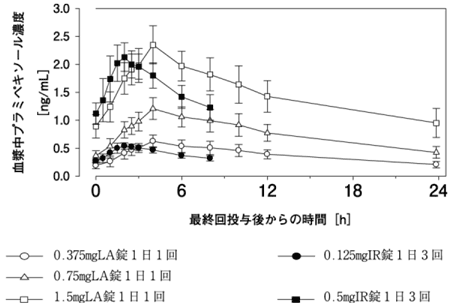
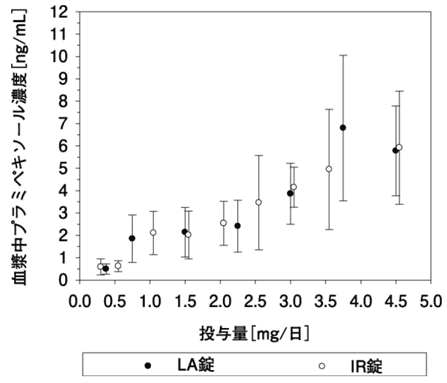
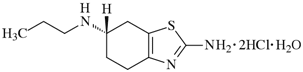
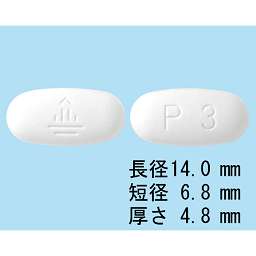
1
Wright C. E. et al: 相互作用に関する試験(2003年12月2日承認、申請資料概要ト.3.(6).3))
2
Yamamura N. et al: 相互作用に関する試験(2003年12月2日承認、申請資料概要ヘ.3.(2))
3
Sha K. et al: 健康成人での薬物動態試験(2011年4月22日承認、CTD 2.7.6.4)
4
Sha K. et al: 国内二重盲検比較試験(2011年4月22日承認、CTD 2.7.6.8)
5
Dansirikul C. et al: 母集団薬物動態解析(2011年4月22日承認、CTD 2.7.2.2.4)
6
Sha K. et al: 生物学的同等性試験(2011年4月22日承認、CTD 2.7.6.12)
7
Yokoyama K. et al:薬物動態. 1999; 14(4):300-308
8
Haeselbarth V. et al: 代謝、排泄に関する試験(2003年12月2日承認、申請資料概要ヘ.3.(1))
9
Salin L. et al: 日本人を含む国際共同試験(2011年4月22日承認、CTD 2.7.6.6)
10
Salin L. et al: 海外国際共同試験(2011年4月22日承認、CTD 2.7.6.7)
11
Mierau J. et al:Eur. J. Pharmacol., 1995; 290:29-36
12
Mierau J.:Clin. Neuropharmacol., 1995; 18:S195-S206
13
Domino E. F. et al:Eur. J. Pharmacol., 1997; 325:137-144
14
Bauer R. et al: 薬効薬理試験(2003年12月2日承認、申請資料概要添付資料ホ-1)
15
Takeuchi S. et al:医学と薬学. 2003; 49(6):973-983

