



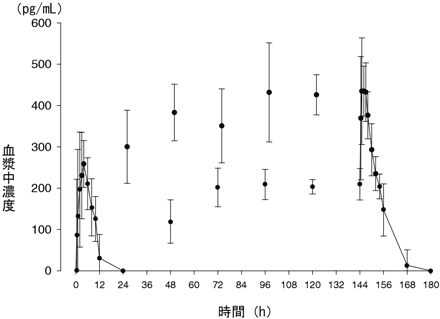
 P6
P6
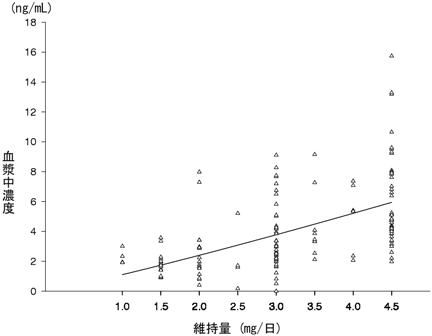
 P8
P8
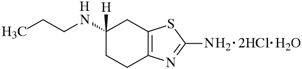
1
入江伸ほか:臨床医薬. 2003;19(2):149-161
2
入江伸ほか:臨床医薬. 2003;19(2):163-177
3
吉川浩一ほか:日本人パーキンソン病患者の薬物動態解析(2003年12月2日承認、申請資料概要ヘ.3.(2))
4
Haeselbarth V. et al:外国健康成人の薬物動態解析(標識)(2003年12月2日承認、申請資料概要ヘ.3.(1))
5
Haeselbarth V. et al:外国健康成人の薬物動態解析(非標識)(2003年12月2日承認、申請資料概要ヘ.3.(1))
6
Sisson L. T. et al:薬物動態に対する食事の影響(2003年12月2日承認、申請資料概要ヘ.3.(1))
7
Yokoyama K et al:薬物動態. 1999;14(4):300-308
8
Wright C. E. et al:外国人腎機能障害患者の薬物動態解析(2003年12月2日承認、申請資料概要ヘ.3.(2))
9
Dansirikul C. et al:母集団薬物動態解析(2010年1月20日承認、CTD 2.7.2.2.4)
10
Wright C. E. et al:薬物動態に対するシメチジンの影響(2003年12月2日承認、申請資料概要ヘ.3.(3))
11
Yamamura N. et al:日本人パーキンソン病患者母集団動態解析(2003年12月2日承認、申請資料概要ヘ.3.(2))
12
Mizuno Y. et al:Movement Disorders. 2003;18(10):1149-1156
13
Shannon K. M.:Neurology. 1997;49(3):724-728
14
Wright C. E. et al:外国人初期パーキンソン病患者第Ⅲ相試験(2003年12月2日承認、申請資料概要ト.3.(4).2))
15
Guttman M.:Neurology. 1997;49(4):1060-1065
16
Sha K. et al:日本人特発性レストレスレッグス症候群患者第Ⅱ相試験(2010年1月20日承認、CTD 2.7.6.1)
17
Sha K. et al:日本人特発性レストレスレッグス症候群患者第Ⅲ相試験(2010年1月20日承認、CTD 2.7.6.2)
18
Mierau J. et al:Eur. J. Pharmacol., 1995;290:29-36
19
Mierau J.:Clin. Neuropharmacol., 1995;18:S195-S206
20
Domino E. F. et al:Eur. J. Pharmacol., 1997;325:137-144
21
Bauer R. et al:作用機序に関する薬理学的検討(2003年12月2日承認、申請資料概要添付資料ホ-1)
22
武内正吾ほか:医学と薬学. 2003;49(6):973-983
23
Ondo WG. et al:Movement Disorders. 2000;15(1):154-158

